Der GLUT1-Defekt ist eine noch sehr „junge“ Erkrankung – er wurde 1991 erstmals beschrieben. Viele Fragen zum Verlauf und zu den vielfältigen Erscheinungen der Erkrankung sind noch unbeantwortet. Hier einige Beispiele: mit steigender Zahl der Patienten werden sich die verschiedenen Formen und Verläufe der Erkrankung besser zuordnen lassen, Langzeiteffekte, Nebenwirkungen und der günstigste Zeitpunkt zum Beenden der ketogenen Diät werden deutlicher werden, ein Tiermodell (GLUT1-knockout Maus) würde wertvolle Informationen über die Erkrankung selbst und den Erfolg neuer Therapien liefern, die Gabe von oralen Ketonen könnte die ketogene Diät optimieren, die Häufigkeit der Erkrankung bei Erwachsenen ist noch völlig unerforscht. Wir stehen somit erst am Anfang, diese Erkrankung zu verstehen und zu behandeln.
Ursache
| Das Gehirn benötigt zur Energiegewinnung Glukose. Diese gelangt über den spezifischen Transporter GLUT1 aus dem Blut in das Gehirn. Ist dieser Transporter defekt, kommt es zu einer Unterversorgung mit Brennstoff, zu einer „Energiekrise“ im Gehirn – eben dem Glukosetransporter(GLUT1)-Defekt. | 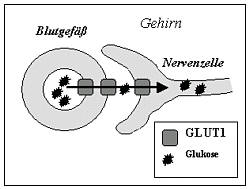 |
Symptome
| Leitsymptom: | zerebrale, meist therapieresistente Krampfanfälle im ersten Lebensjahr, gehäuft nüchtern; Besserung durch Glukosezufuhr• Säuglingsalter: Nickanfälle, Augenverdrehen, komplexe Partialanfälle; keine BNS-Anfälle • Kindesalter: Verlangsamung, "wie betrunken", myoklonische Anfälle, Grand Mal • Erwachsene: Anfallstyp variabel; noch wenig Erfahrung |
| Weitere Leitsymptome: | • erworbener Mikrozephalus • Hypotonie • Ataxie • Dystonie |
Ausblick
- mit steigender Zahl der Patienten werden sich die verschiedenen Formen und Verläufe der Erkrankung besser zuordnen lassen,
- Langzeiteffekte, Nebenwirkungen und der günstigste Zeitpunkt zum Beenden der ketogenen Diät werden deutlicher werden,
- ein Tiermodell (GLUT1-knockout Maus) würde wertvolle Informationen über die Erkrankung selbst und den Erfolg neuer Therapien liefern,
- die Gabe von oralen Ketonen könnte die ketogene Diät optimieren,
- die Häufigkeit der Erkrankung bei Erwachsenen ist noch völlig unerforscht.
Diagnostik
|
Der Quotient (Beispiel):
|
Therapie
Einzige etablierte Therapie des GLUT1-Defektes ist derzeit eine sogenannte ketogene Ernährungstherapie (KET). Dabei handelt es sich um eine extrem fettreiche, kohlenhydratarme, isokalorische Diät, die das Fasten imitiert.
Im Fasten wird Körperfett zu Energie verbrannt; es entstehen Ketone, die im Gehirn Glukose als Energieträger nahezu vollständig ersetzen können. Die ketogene Ernährung stellt Nahrungsfett bereit und verhindert so den Abbau von Körperfett, ohne dass sich an der Stoffwechselsituation des Fastens etwas ändert. Die entstehenden Ketone gelangen mittels eines separaten Transportersystems in das Gehirn. Sie dienen ihm als alternative Energiequelle und können so den Glukosemangel weitgehend ausgleichen.
Die ketogene Ernährungstherapie darf nur unter ärztlicher Aufsicht durchgeführt werden; Vitamine, Mineralstoffe und Spurenelemente müssen ggf. zugeführt werden, der hohe Fettanteil muss in jeder Mahlzeit eingehalten werden.
Der Erfolg dieser Behandlung ist beeindruckend: fast alle Patienten werden innerhalb weniger Wochen anfallsfrei, ihre Entwicklung zeigt deutliche Fortschritte.
Zahlreiche Substanzen und Medikamente hemmen den GLUT1-Transporter (darunter Coffein, Phenobarbital, Narkotika, Chloralhydrat) oder zeigen unerwünschte Wechselwirkungen mit der ketogenen Diät (Valproat, Topiramat, Acetazolamid) und sollten bei dieser Erkrankung vermieden werden.
Ausblick
- mit steigender Zahl der Patienten werden sich die verschiedenen Formen und Verläufe der Erkrankung besser zuordnen lassen,
- Langzeiteffekte, Nebenwirkungen und der günstigste Zeitpunkt zum Beenden der ketogenen Diät werden deutlicher werden,
- ein Tiermodell (GLUT1-knockout Maus) würde wertvolle Informationen über die Erkrankung selbst und den Erfolg neuer Therapien liefern,
- die Gabe von oralen Ketonen könnte die ketogene Diät optimieren,
- die Häufigkeit der Erkrankung bei Erwachsenen ist noch völlig unerforscht.