Glukosetransporter(GLUT1)-Defekt
der Glukosetransporter(GLUT1)-Defekt
Der GLUT1-Defekt ist eine noch sehr „junge“ Erkrankung – er wurde 1991 erstmals beschrieben.
Er ist eine der wenigen stoffwechselbedingten, aber gut behandelbaren Epilepsien im Kindesalter – daher sollte diese Erkrankung bei allen unklaren Epilepsien im Kindesalter ausgeschlossen werden!
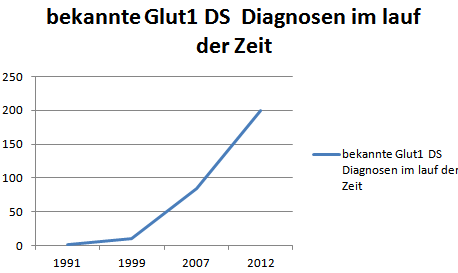
nach Klepper 2014
Weltweit mehr als 200 Patienten bekannt.
Viele Fragen zum Verlauf und zu den vielfältigen Erscheinungen der Erkrankung sind noch unbeantwortet.
Hier einige Beispiele:
* mit steigender Zahl der Patienten werden sich die verschiedenen Formen und Verläufe der Erkrankung besser zuordnen lassen,
* Langzeiteffekte, Nebenwirkungen und der günstigste Zeitpunkt zum Beenden der ketogenen Diät werden deutlicher werden,
* ein Tiermodell (GLUT1-knockout Maus) würde wertvolle Informationen über die Erkrankung selbst und den Erfolg neuer Therapien liefern,
* die Gabe von oralen Ketonen könnte die ketogene Diät optimieren,
* die Häufigkeit der Erkrankung bei Erwachsenen ist noch völlig unerforscht.
Wir stehen somit erst am Anfang, diese Erkrankung zu verstehen und zu behandeln.
Ursache
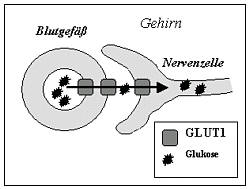
Das Gehirn benötigt zur Energiegewinnung Glukose. Diese gelangt über den spezifischen Transporter GLUT1 aus dem Blut in das Gehirn.
Ist dieser Transporter defekt, kommt es zu einer Unterversorgung mit Brennstoff (Hypoglykorrhachie), zu einer „Energiekrise“ im Gehirn – eben dem Glukosetransporter(GLUT1)-Defekt.
Häufigste Ursache des Transporter-Defekts ist eine Mutation am SLC2A1-Gen (OMIM 138140, Genkartenlokus 1p35-31.3)
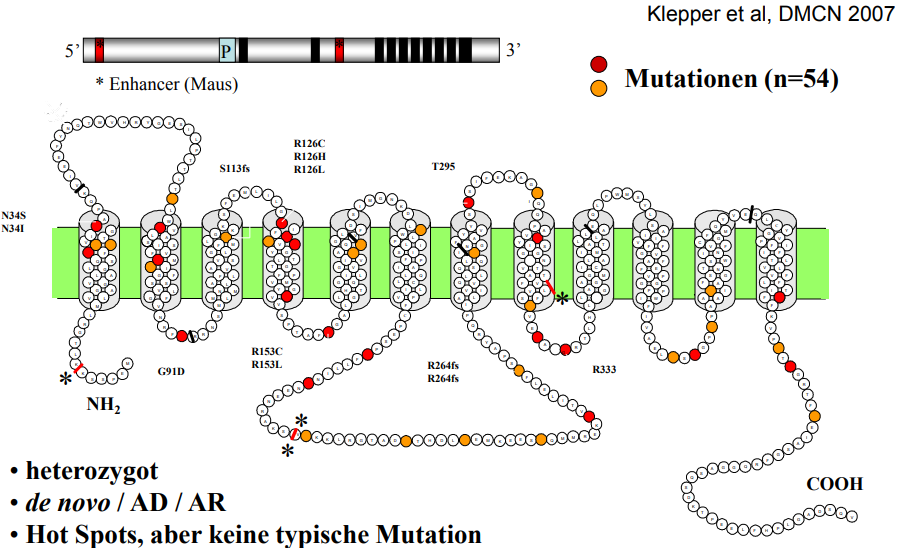
Der Phänotyp ist sehr variabel und mehrere atypische Varianten wurden beschrieben. (siehe Symptome)
(vgl. Klepper in Epliepsia 2008 ( https://www.ncbi.nlm.nih.gov/pubmed/19049586 ); Klepper 2014 )
Symptome
| Leitsymptom: | zerebrale, meist therapieresistente Krampfanfälle im ersten Lebensjahr, gehäuft nüchtern; Besserung durch Glukosezufuhr• Säuglingsalter: Nickanfälle, Augenverdrehen, komplexe Partialanfälle; keine BNS-Anfälle • Kindesalter: Verlangsamung, "wie betrunken", myoklonische Anfälle, Grand Mal • Erwachsene: Anfallstyp variabel; noch wenig Erfahrung |
| Weitere Leitsymptome: | • erworbener Mikrozephalus • Hypotonie • Ataxie • Dystonie |
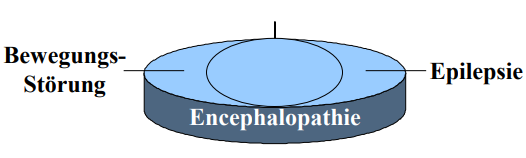
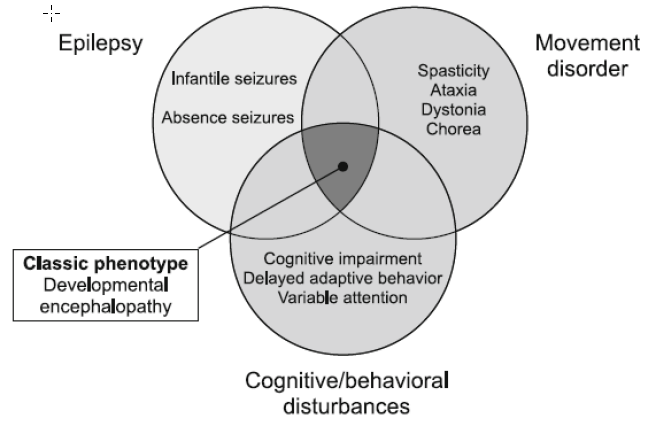
Zudem manifestiert sich die phänotypische Ausprägung des Glut1 Defekts in einer breiten Varianz, die sich Spannungsfeld zwischen Bewegungsstörung und Epilepsie ausprägt.
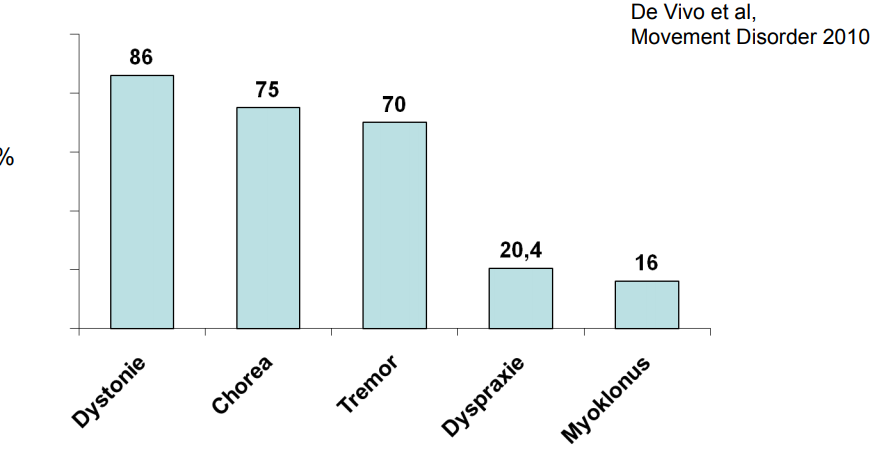
Es werden laut Literatur(1) verschiedene Bewegungsstörungen bei Patienten mit Glut1 Defekt beobachtet. Wie Dystonie, Chorea, Tremor, Dyspraxie und Myoklonus. Die Ausprägung ist dabei sehr individuell, scheint jedoch um so stärker ausgeprägt zu sein, je später der Glut1 Defekt erkannt und adäquat behandelt wird.
Im erweiterten Phänotyp werden zudem weitere Arten von Bewegungstörungen wie Paroxysmal oder PEDystonie unterschieden.
Folgende Übersicht gibt eine Übersicht, wie häufig diverse Bewegungsstörungen beobachetet wurden.(1)
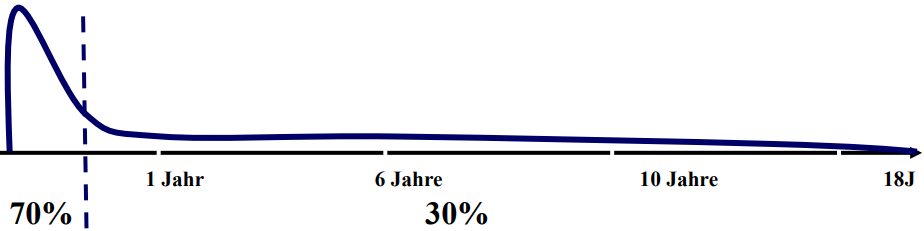
Auch bei der epileptischen Ausprägung des Glut1 Defekts sind variable, oft therapieresistente, Anfallstypen zu beobachten. Wie:
- Zyanose-Attacken
- Absencen
- fokal / generalisiert Anfälle
- Myoclonisch-astatisch
In der Literatur scheint es zudem eine Beobachtungshäufung von Epilepsie i.v.m dem Glut1 Defekt im Kleinkindalter zu geben.
Forschungsansätze
Der GLUT1-Defekt ist eine seltene Erkrankung – er wurde 1991 erstmals beschrieben. Viele Fragen zum Verlauf und zu den vielfältigen Erscheinungen der Erkrankung sind noch unbeantwortet. Hier einige Beispiele:
- mit steigender Zahl der Patienten werden sich die verschiedenen Formen und Verläufe der Erkrankung besser zuordnen lassen,
- Langzeiteffekte, Nebenwirkungen und der günstigste Zeitpunkt zum Beenden der ketogenen Diät werden deutlicher werden,
- ein Tiermodell (GLUT1-knockout Maus) würde wertvolle Informationen über die Erkrankung selbst und den Erfolg neuer Therapien liefern,
- die Gabe von oralen Ketonen könnte die ketogene Diät optimieren,
- die Häufigkeit der Erkrankung bei Erwachsenen ist noch völlig unerforscht.
Wir stehen somit erst am Anfang, diese Erkrankung zu verstehen und zu behandeln.
Diagnostik
|
Der Quotient (Beispiel):
|
Entscheidend ist das Verhältnis von Liquorglukose zu Blutglukose (s.o.): normalerweise transportiert GLUT1 ca. 60% der Blutglukose in das Gehirn (Quotient 0,6). Beim GLUT1-Defekt ist die Liquorglukose und damit der Quotient deutlich niedriger (< 0,4). Der Quotient gibt nur Auskunft über das Vorliegen, nicht jedoch über die Schwere der Erkrankung.
Die zerebrale Bildgebung (Kernspin = MRT, Computertomogramm = CT) ist in der Regel unauffällig.
Ist die Diagnose durch den Nachweis von wiederholt niedriger Liquorglukose und niedrigem Quotienten bestätigt, kann die Anzahl und die Funktion des GLUT1-Transporters in roten Blutkörperchen bestimmt werden. Diese aufwändigen Untersuchungen sind Speziallabors vorbehalten.
Das Gen für GLUT1 ist bekannt: es befindet sich auf dem kurzen Arm von Chromosom 1 (1p35-31.3). Es lässt sich daher auf Mutationen untersuchen. Hierfür genügt eine einfache Blutentnahme (EDTA-Blut) – die Diagnostik erfolgt ebenfalls in Speziallabors.
Mittlerweile sind zahlreiche Mutationen bei Patienten mit GLUT1-Defekt nachgewiesen.
Therapie
Einzige etablierte Therapie des GLUT1-Defektes ist derzeit eine sogenannte ketogene Diät. Dabei handelt es sich um eine extrem fettreiche, kohlenhydratarme, isokalorische Diät, die das Fasten imitiert.
Im Fasten wird Körperfett zu Energie verbrannt; es entstehen Ketone, die im Gehirn Glukose als Energieträger nahezu vollständig ersetzen können. Die ketogene Diät stellt Nahrungsfett bereit und verhindert so den Abbau von Körperfett, ohne dass sich an der Stoffwechselsituation des Fastens etwas ändert. Die entstehenden Ketone gelangen mittels eines separaten Transportersystems in das Gehirn. Sie dienen ihm als alternative Energiequelle und können so den Glukosemangel weitgehend ausgleichen.
Die ketogene Diät darf nur unter ärztlicher Aufsicht durchgeführt werden; Vitamine, Mineralstoffe und Spurenelemente müssen zugeführt werden, der hohe Fettanteil muss in jeder Mahlzeit eingehalten werden.
Der Erfolg dieser Behandlung ist beeindruckend: viele Patienten werden innerhalb weniger Wochen anfallsfrei, ihre Entwicklung zeigt deutliche Fortschritte.
Zahlreiche Substanzen und Medikamente hemmen den GLUT1-Transporter (darunter Coffein, Phenobarbital, Narkotika, Chloralhydrat) oder zeigen unerwünschte Wechselwirkungen mit der ketogenen Diät (Valproat, Topiramat, Acetazolamid) und sollten bei dieser Erkrankung vermieden werden.