
Alles zu Glut1DS
Glukosetransporterdefizitsyndrom
- Seit 2021 hat der Glut1DS mit der Bezeichnung E74.810 auch einen eigenen Klassifikationscode im ICD-10.
- Bereits 2002 haben betroffene Familien den Förderverein Glukosetransporter (Glut1)-Defekt e.V. gegründet und Herrn Prof. Dr. Klepper als wissenschaftlichen Berater gewonnen.
- 1999 wurde mit der Forschung durch Herrn Prof. Dr. Jörg Klepper in Deutschland begonnen.
- Das Glukosetransporterdefizitsyndrom (Glut1DS) wurde erstmals 1989 von Herrn Prof. Darryl De Vivo in den USA beschrieben.
- Der Glukosetransporter Typ 1-Defekt wurde erstmals 1989 von Herrn Prof. Darryl De Vivo in den USA beschrieben.
- 1999 wurde mit der Forschung durch Herrn Prof. Dr. Jörg Klepper in Deutschland begonnen.
- Bereits 2002 haben betroffene Familien den Förderverein Glukosetransporter (Glut1)-Defekt e.V. gegründet und Herrn Prof. Dr. Klepper als wissenschaftlichen Berater gewonnen.
- Seit 2021 hat der Glut1DS mit der Bezeichnung E74.810 auch einen eigenen Klassifikationscode im ICD-10.
Viele Fragen zum Verlauf und zu den vielfältigen Erscheinungen der Erkrankung sind noch unbeantwortet. Hier einige Beispiele:
Wie häufig tritt die Erkrankung auf? Wie kann eine möglichst frühe Diagnose umgesetzt werden?
Gibt es Langzeitfolgen durch die ketogene Ernährungstherapie (KET)? Kann die KET irgendwann beendet werden?
Welche Auswirkungen hat die KET auf eine Schwangerschaft?
Kann die Gabe von oralen Ketonen die KET optimieren?
Wie kann das Tiermodell (Glut1DS-knockout Maus) wertvolle Informationen über die Erkrankung selbst und den Erfolg neuer Therapien beitragen?
Langzeiteffekte, Nebenwirkungen und ob bzw. wann die ketogene Ernährungs-therapie beendet werden, kann werden deutlicher!
Mit steigender Zahl der Betroffenen werden sich die verschiedenen Formen und Verläufe der Erkrankung besser zuordnen lassen.
Die Gabe von oralen Ketonen könnte die ketogene Ernährungstherapie optimieren!
Kann die Gabe von oralen Ketonen die KET optimieren?
Wie kann das Tiermodell (Glut1DS-knockout Maus) wertvolle Informationen über die Erkrankung selbst und den Erfolg neuer Therapien beitragen?
Wir stehen somit erst am Anfang, diese Erkrankung zu verstehen und zu behandeln. Mit steigender Zahl der Betroffenen werden sich die verschiedenen Formen und Verläufe besser zuordnen lassen.
Was ist Glut1DS?
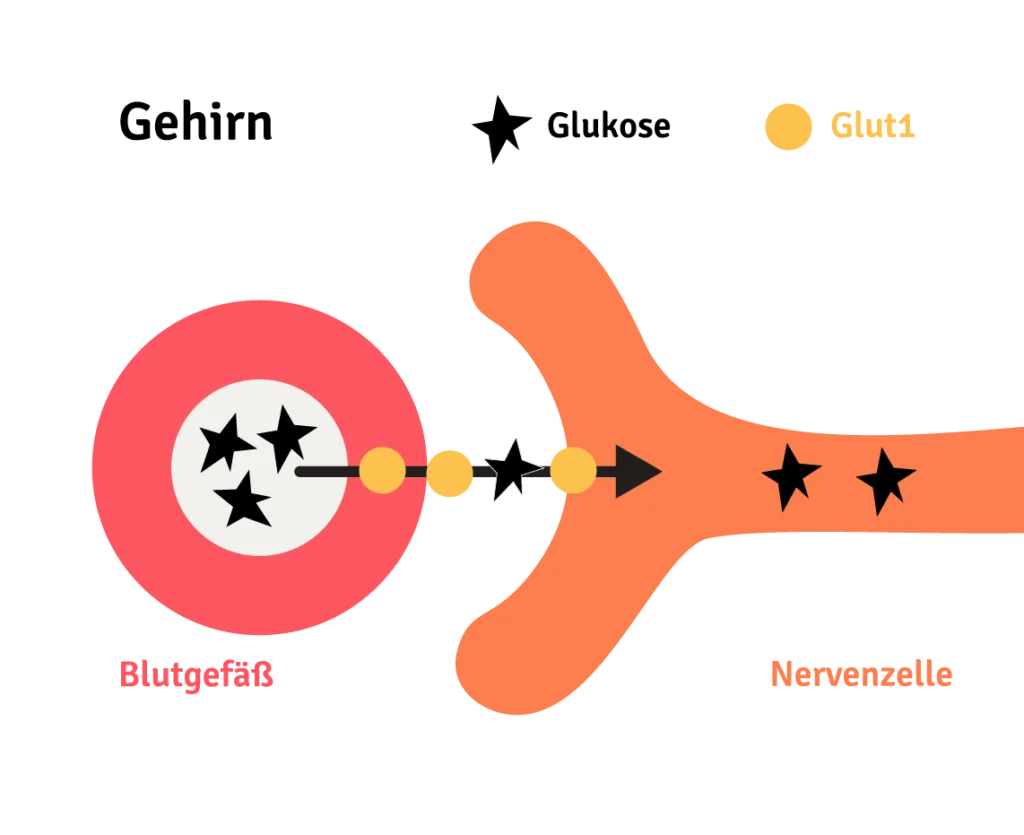
Normalerweise sorgt der Glukose-Transporter-Typ 1 (Glut1) dafür, dass Glukose als Energieträger für den Hirnstoffwechsel aus dem Blut ins Gehirn transportiert wird. Wenn der Glut1-Transporter defekt ist, kann er diese Aufgabe nicht richtig erfüllen und es kommt im Gehirn zu einem Energiemangel. Das Glukose-Transporter-Typ-1-Defizit-Syndrom (Glut1DS) auch bekannt als Glukosetransporter-Defekt, G1D, De Vivo-Krankheit ist daher eine Stoffwechselerkrankung und zählt zu den seltenen Erkrankungen. Wir im Verein sprechen auch oft einfach nur von Glut1. Die Ursache für Glut1DS ist eine genetische Veränderung, die durch Mutation des SLC2A1-Gens entweder neu entsteht (de novo) oder von einem der Elternteile vererbt wird. Die Auswirkungen sowie die Behandlung sind in diesem 2-seitigen Informationsblatt für Betroffene und Betreuungspersonen bündig zusammengefasst. Hier geht es zum Flyer in leichter Sprache.

Therapie
Im Moment gibt es für den Glut1DS keine Heilung und keine Behandlung mit Medikamenten. Die Standardtherapie ist die ketogene Ernährungstherapie (KET). Dabei wird die Fettmenge in der Nahrung erhöht und gleichzeitig Kohlenhydrate und Eiweiß (Protein) reduziert, wodurch der Körper von Kohlenhydratstoffwechsel auf Fettstoffwechsel umstellt. Durch die ketogene Ernährung können bestimmte Symptome vom Glut1DS unter Umständen ganz verhindert oder ihr Schweregrad verändert werden, was die Gesamtentwicklung positiv beeinflusst. Bei diesem Fettstoffwechsel entstehen bei der Verarbeitung von Fett in der Leber Ketonkörper. Statt der Glukose, die durch den defekten Glut1-Transporter vor allem im Gehirn nicht ausreichend zur Verfügung steht, können Ketonkörper als alternative Energiequelle die Versorgung von Gehirn und auch Körper übernehmen. Sie nutzen einen anderen und funktionierenden „Transportweg“, der die Blut-Hirn-Schranke passieren kann. Auch wenn die KET herausfordernd ist, ist sie für die meisten Betroffenen die aktuell beste Therapie: Es wird empfohlen die KET so früh wie möglich zu beginnen. Auf unserer Seite „Für Ketarier“ wird die KET ausführlich dargestellt.
Was sind die Symptome und wann treten sie auf?
Die Folgen vom Glut1DS können ganz unterschiedlich sein: von milden Verläufen z. B. geringerer Leistungsfähigkeit, über Absencen und Krampfanfällen (Epilepsie) bis hin zu teils weitreichenden geistigen und körperlichen Einschränkungen. Keines der Symptome ist eindeutig und kommt immer vor, daher kann Glut1DS auch nicht einfach erkannt werden.
Manche Betroffenen haben schon als Säuglinge in den ersten Lebenswochen Symptome, bei anderen zeigen sie sich erstmals, wenn sie Kinder sind, bei anderen fast überhaupt nicht, sodass manche unter Umständen bis ins Erwachsenenalter nicht wissen, dass sie betroffen sind. Mit zunehmendem Alter verändern sich die Symptome auch, sodass Säuglinge, Kinder, Jugendliche und Erwachsene andere Symptome haben, und diese in unterschiedlichen Schweregraden, obwohl die Ursache dieselbe ist.
Weil es immer noch sehr wenige Betroffene gibt, und bei diesen die Symptome und ihre Ausprägung sehr unterschiedliche sein können, gibt es keinen eindeutigen Katalog, sondern ein Zusammenspiel unterschiedlicher Merkmale, die gemeinsam erst das Bild ergeben. Einige davon werden im Folgenden dargestellt.

Epilepsien
Epilepsien zählen zu den ersten und häufigsten äußeren Anzeichen für Glut1DS. Sie treten als frühkindliche Epilepsien schon im Säuglingsalter oder in der frühen Kindheit (vor 4 Jahren) auf und kommen mit zunehmendem Alter seltener vor. Dabei ist auch hier die Bandbreite der Ausprägung sehr unterschiedlich: in manchen Fällen sind sogenannte Absencen zu beobachten, bei denen das Kind für einige Momente abwesend wirkt. Bei anderen kommt es zu größeren Krampfanfällen mit Muskelzuckungen (Myoklonien), dem Verlust der Muskelspannung (atonisch) bis hin zum Sturz und kurzzeitiger Bewusstlosigkeit. Die Art dieser Anfälle kann unterschiedlich sein und auf den ersten Blick mit anderen Formen der Epilepsie verwechselt werden. Sie sind auch bei der Elektroenzephalographie – EEG- (Messung der Hirnströme) nicht eindeutig als Folge von Glut1DS zu erkennen. In der Regel zeigen Medikamente gegen Epilepsie (Antikonvulsiva) keine Wirkung, was oft ein starker Hinweis auf Glut1DS ist.
In manchen Fällen aber helfen die Medikamente gegen die Anfälle, doch andere Symptome bleiben oder kommen hinzu. Dadurch dauert es in der Regel in solchen Fällen länger bis zur Diagnose von Glut1DS.
Mit Beginn einer ketogenen Ernährungstherapie kann schon innerhalb von zwei Tagen eine Anfallsreduzierung bis hin zur Anfallsfreiheit erreicht werden, und nach stabiler Einführung können oft zuvor eingesetzte Medikamente abgesetzt werden. Manche Betroffene müssen allerdings weiterhin zusätzlich Medikamente einnehmen. In manchen Fällen aber helfen die Medikamente gegen die Anfälle, doch andere Symptome bleiben oder kommen hinzu. Dadurch dauert es in der Regel in solchen Fällen länger bis zur Diagnose von Glut1DS.
Bewegungsstörungen
In der Regel deuten Epilepsien, die nicht oder nicht gut mit Medikamenten behandelt werden können, in Kombination mit Bewegungsstörungen auf Glut1DS hin.
Bewegungsstörungen (Ataxie) sind das zweithäufigste Anzeichen nach den Krampfanfällen und gelten als charakteristisch für Glut1DS. Vor allem bei Kleinkindern sind es oft plötzlich und nur kurzzeitig auftretende schnelle ruckartige Augenbewegungen, häufig begleitet von einem leichten Drehen des Kopfes in die gleiche Richtung, bei denen das Kind nicht abwesend oder eingetrübt wirkt (paroxysmale Augen-Kopf-Bewegung).
In der Folge und mit zunehmendem Alter werden diese Bewegungsstörungen bei manchen Betroffenen komplexer und sind als allgemeine Ungeschicklichkeit bis hin zu starken Auffälligkeiten in den Bewegungsabläufen zu beobachten: z.B. unwillkürliche spontane Bewegungen, plötzliche Schwäche, Kurzatmigkeit oder auch Sprachstörungen in unterschiedlich starken Ausprägungen. Anfallsartige Bewegungsstörungen können nach körperlicher Belastung (paroxysmale belastungsinduzierte Dyskinesie, PED) auftreten. Andere Auslöser für Bewegungsstörungen sind emotionaler Stress, Fieber, Erschöpfung, unzureichende Ketose, Schlafentzug, Temperaturschwankungen und Drogen. Bei den Bewegungsstörungen kann die Bandbreite ebenfalls von sehr ernst bis eher unauffällig reichen. Auch hier kann die ketogene Ernährungstherapie in der Regel sofort positiv einwirken und die Störungen verschwinden oder werden gemildert.
Kognitive Einschränkungen und Sprache
Kognitive Einschränkungen sind ein häufiges Symptom dieser Erkrankung, da das Gehirn auf Glukose als Hauptenergiequelle angewiesen ist. Zu den möglichen kognitiven Beeinträchtigungen zählen Lernschwierigkeiten, Gedächtnisprobleme und allgemeine Entwicklungsverzögerungen. Eine frühzeitige Diagnose und Intervention sind entscheidend, um die kognitiven Fähigkeiten bestmöglich zu fördern.
Der Glut1DS kann auch Sprachstörungen verursachen, da die betroffene Person Schwierigkeiten mit der neurologischen Verarbeitung von Sprache haben kann. Sprachentwicklungsverzögerungen sind bei vielen Kindern mit dieser Erkrankung zu beobachten. Die Ursache hierfür liegt oft in den allgemeinen kognitiven Einschränkungen und den Herausforderungen in der Sprachwahrnehmung und -produktion.
Zusätzlich können auch motorische Probleme, die durch den Glut1DS hervorgerufen werden, die Artikulation und die Sprachmotorik beeinträchtigen.
Weitere Symptome
Oft geht Glut1DS mit einer globalen Entwicklungsverzögerung sowie Weiteren mehr oder weniger ausgeprägten und selten Symptomen einher. Dazu gehören, z. B.
• ein verlangsamtes Kopfwachstum im Vergleich zu Kindern der gleichen Altersgruppe,
• halbseitige Lähmungen,
• nächtliche Muskelkrämpfe in den Beinen,
• und einige andere.
Wie wird Glut1DS festgestellt?
In der Regel werden eine oder mehrere der folgenden Untersuchungen durchgeführt, wenn andere Erkrankungen ausgeschlossen wurden und mehrere der beschriebenen Symptome zusammen auftreten.
EEG-Messung
Bei der Elektroenzephalografie (EEG) werden die elektrischen Aktivitäten im Gehirn gemessen. Das Verfahren wird sehr oft bei Epilepsien angewendet. Bei Glut1DS aller Altersgruppen ist ein EEG, das zwischen den Anfällen erstellt wird, oft normal. Auffälligkeiten treten in bestimmten Altersgruppen häufiger auf: Bei Säuglingen sind fokale Verlangsamungen und epilepsieartige Entladungen häufiger, während bei Kindern ab zwei Jahren ein Spike-Wave-Muster zu beobachten ist. Bei Menschen mit Glut1DS ohne ketogene Ernährungstherapie (KET) kann ein Merkmal sein, dass sich EEG-Auffälligkeiten vor dem Essen mit der Nahrungsaufnahme verbessern, wenn dem unterversorgten Gehirn wieder Glukose zugeführt wird.
Da es bei der Diagnose ebenfalls kein wirklich eindeutiges Merkmal gibt, ist es wichtig alle Informationen, einschließlich der Symptome und Reaktionen auf KET und Medikamente ganzheitlich zu betrachten.
Lumbalpunktion
Bei der Lumbalpunktion wird unter Betäubung Nervenwasser (Liquor) aus dem Wirbelsäulenkanal entnommen. Entscheidend ist das Verhältnis von Liquorglukose zu Blutglukose. Normalerweise transportiert der Glut1-Transporter ca. 60% der Blutglukose in das Gehirn (Quotient 0,6). Beim Glut1DS ist die Liquorglukose und damit der Quotient deutlich niedriger (< 0,4). Der Quotient gibt nur Auskunft über das Vorliegen, nicht jedoch über die Schwere der Erkrankung. Die Blutzuckermessung sollte unmittelbar vor der Lumbalpunktion durchgeführt werden, um eine stressbedingte Beeinträchtigung zu vermeiden. Bei Glut1DS sind die Laktatwerte im Liquor ebenfalls immer niedriger als normal oder abnormal niedrig, was diesen Zustand von anderen Erkrankung unterscheidet.
Genetischer Test
Über eine einfache Blutentnahme kann von Speziallaboren eine Genanalyse durchgeführt werden. Diese zeigen bei 81 bis 89% der Betroffenen Veränderungen auf dem SLC2A1 Gen. Wie die Werte zeigen, gilt das nicht für alle Betroffenen. Glut1DS kann auch vorliegen, ohne dass eine Veränderung des Gens festzustellen ist. Die Art der genetischen Mutation ist häufig mit dem Schweregrad der Symptome verbunden: Missense-Varianten (leichter und mittlerer Schweregrad); Spleißstellen- und Nonsense-Varianten sowie Insertionen, Deletionen und Exon-Deletionen (mittlerer und schwerer Schweregrad); und komplette Gen-Mikrodeletionen (schwerer Schweregrad).
Wer forscht daran?
Weltweit gibt es Zentren, die gut miteinander vernetzt sind und sich über neueste Forschungsergebnisse regelmäßig auf Fachtagungen austauschen. Im Jahr 2023 wurde ein Consensus-Paper von Klepper et al. veröffentlicht, dass aus den Forschungsergebnissen kooperierender Ärztinnen und Ärzte weltweit zusammengetragen wurde. Dieser Artikel stellt den aktuellen Kenntnisstand dar.
In Deutschland gibt es an einigen Kliniken spezialisierte Ärztinnen und Ärzte, die in der Behandlung und Begleitung von Kindern mit Glut1DS sehr erfahren sind. Da Glut1DS zu den seltenen Erkrankungen zählt sind niedergelassene Kinderärzte nicht im gleichen Maße informiert und kompetent. Sie stellen aber die normale Versorgung der Kinder sicher und informieren sich in der Regel, um Glut1DS Betroffenen behandeln zu können.
Durch die bessere Diagnose bei Kindern in den letzten Jahren, steigt jetzt auch die Anzahl der erwachsenen Betroffenen und damit auch die Erfahrung bei der Symptomveränderung und der Ernährungstherapie. Allerdings ist das auch eine große Herausforderung, da viele Betroffene von der kinderärztlichen Betreuung in die Erwachsenenmedizin wechseln. Diese Transition ist schwierig, weil es kaum Ärzte gibt, die sich mit Glut1DS auskennen, das gilt für Allgemeinärzte ebenso wie z. B. für neurologische Fachärzte.
Eine Checkliste für die Transition gibt’s hier!
Wenn ihr auf medizinisches Personal trefft, die Glut1DS noch nicht kennen, dann empfehlen wir euch, dieses 1-seitige Informationsblatt für Gesundheitsfachkräfte zusammen mit der Darstellung eines klassischen Patientenweges weiterzugeben. Insbesondere der Umfang der Verlaufskontrollen kann dieser 1-seitigen Übersicht schnell entnommen werden.
Downloads/Links zu diesem Thema
Jetzt die Liste mit Ärztinnen und Ärzten, die Menschen mit Glut1DS betreuen, bei uns anfragen!
Oft gestellte Fragen
Was ist Glut1DS?
Das Glukose-Transporter-Typ-1-Defizit-Syndrom (Glut1DS) ist eine genetisch bedingte Stoffwechselerkrankung und zählt zu den seltenen Erkrankungen. Weil der Glukosetransporter defekt ist, kommt Glukose nicht in ausreichender Menge aus dem Blut im Gehirn an. Das hat zur Folge, dass Menschen mit Glut1DS Epilepsien, Bewegungs- und Entwicklungsstörungen haben können.
Wie erkennt man Glut1DS?
Für Glut1DS gibt es kein einfaches, eindeutiges Merkmal. In der Regel haben Menschen mit Glut1DS durch die Unterversorgung mit Glukose z.B. Epilepsien, die nicht gut mit Medikamenten behandelt werden können, sowie Bewegungsstörung und kognitive Einschränkungen. Diese Symptome können aber auch bei anderen Erkrankungen auftreten. Hinzukommt, dass Symptome bei einigen Betroffenen schon im Säuglingsalter, bei anderen erst später auftreten und sie zudem mehr oder weniger ausgeprägt sind. Wenn diese Symptome auftreten, kann man die Erkrankung heute gut durch einen Gentest oder eine Lumbalpunktion feststellen.
Welche Behandlung gibt es für Glut1DS?
Die Therapie der Wahl bei Glut1DS ist die ketogene Ernährungstherapie. Dafür wird jede Mahlzeit so berechnet, dass der Anteil von Fett wesentlich höher ist als der Anteil von Kohlenhydraten und Eiweiß. Durch den Überschuss an Fett und den Mangel an Kohlenhydraten, stellt der Körper auf einen Keton-Stoffwechsel um, und nutzt die dadurch gebildeten Ketonkörper als alternative Energiequelle. Dadurch sind die Betroffenen wieder mit Energie versorgt, können sich besser entwickeln und sind oft leistungsfähiger.
Für immer ketogen?
Da das Glukose-Transporter-Typ-1-Defizit-Syndrom (Glut1DS) genetisch bedingt ist, verschwindet es nicht. Die ketogene Ernährungstherapie ist im Moment die einzige wirksame Behandlung. Vor allem in der Wachstumsphase bis Mitte zwanzig ist sie sehr wichtig, weil das Gehirn und der Körper in dieser Zeit sehr viel Energie für die Entwicklung brauchen. Mit erwachsenen Betroffenen hat man bisher sehr wenig Erfahrung, aber man geht davon aus, dass auch für diese Menschen eine Ernährung mit wenig Kohlenhydraten sinnvoll ist, möglicherweise aber eine andere Form gewählt werden kann.
Zu welchen Ärzten gehe ich mit Glut1DS?
Die erste Anlaufstelle sind immer Kinderärzte. Wenn der Verdacht für Glut1DS vorliegt, sind Neurologen und im nächsten Schritt Humangenetiker zur Diagnose üblich. Nach der Diagnose ist die beste Anlaufstelle ein spezialisiertes Sozialpädiatrisches Zentrum (SPZ), das sich mit der Behandlung von Menschen mit Glut1DS gut auskennt. In der Regel läuft die Behandlung aller Themen rund um Glut1DS dann über das SPZ und die Kinderärzte zu Hause begleiten das Kind weiterhin wegen aller anderen Erkrankungen, die uns im Leben begegnen. Erwachsene Menschen mit Verdacht auf Glut1DS können sich an die Zentren für Seltene Erkrankungen wenden. Eine Liste mit spezialisierten Zentren und Ärztinnen und Ärzten hat der Glut1-Verein.
Glut1DS und Schwangerschaft?
Menschen mit Glut1DS können Nachwuchs zeugen. Die Wahrscheinlichkeit, dass das Kind eines Menschen mit Glut1DS und eines Menschen ohne Glut1DS ebenfalls Glut1DS bekommt ist 50:50. In der Fachliteratur gibt es eine Veröffentlichung von van der Louw et al. (2017) über eine Fallserie über Mütter, die sich auf Grund von Epilepsie ketogen ernähren und mögliche Einflüsse auf die Schwangerschaft. In einem weiteren Fachartikel von Kramer und Smith (2021) stellt einen Fallbericht einer Mutter mit Glut1DS dar, die schwanger wurde und deren Kind ebenfalls mit dem Glut1DS geboren wurde. Die Mutter ernährte sich auch während der Schwangerschaft ketogen und das Kind ab dem Tag der Geburt. Das Kind ist bisher altersgerecht entwickelt und ist symptomfrei.
Was ist Glut1DS?
Das Glukose-Transporter-Typ-1-Defizit-Syndrom (Glut1DS) ist eine genetisch bedingte Stoffwechselerkrankung und zählt zu den seltenen Erkrankungen. Weil der Glukosetransporter defekt ist, kommt Glukose nicht in ausreichender Menge aus dem Blut im Gehirn an. Das hat zur Folge, dass Menschen mit Glut1DS Epilepsien, Bewegungs- und Entwicklungsstörungen haben können.
Wie erkennt man Glut1DS?
Für Glut1DS gibt es kein einfaches, eindeutiges Merkmal. In der Regel haben Menschen mit Glut1DS durch die Unterversorgung mit Glukose z.B. Epilepsien, die nicht gut mit Medikamenten behandelt werden können, sowie Bewegungsstörung und kognitive Einschränkungen. Diese Symptome können aber auch bei anderen Erkrankungen auftreten. Hinzukommt, dass Symptome bei einigen Betroffenen schon im Säuglingsalter, bei anderen erst später auftreten und sie zudem mehr oder weniger ausgeprägt sind. Wenn diese Symptome auftreten, kann man die Erkrankung heute gut durch einen Gentest oder eine Lumbalpunktion feststellen.
Welche Behandlung gibt es für Glut1DS?
Die Therapie der Wahl bei Glut1DS ist die ketogene Ernährungstherapie. Dafür wird jede Mahlzeit so berechnet, dass der Anteil von Fett wesentlich höher ist als der Anteil von Kohlenhydraten und Eiweiß. Durch den Überschuss an Fett und den Mangel an Kohlenhydraten, stellt der Körper auf einen Keton-Stoffwechsel um, und nutzt die dadurch gebildeten Ketonkörper als alternative Energiequelle. Dadurch sind die Betroffenen wieder mit Energie versorgt, können sich besser entwickeln und sind oft leistungsfähiger.
Für immer ketogen?
Da das Glukose-Transporter-Typ-1-Defizit-Syndrom (Glut1DS) genetisch bedingt ist, verschwindet es nicht. Die ketogene Ernährungstherapie ist im Moment die einzige wirksame Behandlung. Vor allem in der Wachstumsphase bis Mitte zwanzig ist sie sehr wichtig, weil das Gehirn und der Körper in dieser Zeit sehr viel Energie für die Entwicklung brauchen. Mit erwachsenen Betroffenen hat man bisher sehr wenig Erfahrung, aber man geht davon aus, dass auch für diese Menschen eine Ernährung mit wenig Kohlenhydraten sinnvoll ist, möglicherweise aber eine andere Form gewählt werden kann.
Zu welchen Ärzten gehe ich mit Glut1DS?
Die erste Anlaufstelle sind immer Kinderärzte. Wenn der Verdacht für Glut1DS vorliegt, sind Neurologen und im nächsten Schritt Humangenetiker zur Diagnose üblich. Nach der Diagnose ist die beste Anlaufstelle ein spezialisiertes Sozialpädiatrisches Zentrum (SPZ), das sich mit der Behandlung von Menschen mit Glut1DS gut auskennt. In der Regel läuft die Behandlung aller Themen rund um Glut1DS dann über das SPZ und die Kinderärzte zu Hause begleiten das Kind weiterhin wegen aller anderen Erkrankungen, die uns im Leben begegnen. Erwachsene Menschen mit Verdacht auf Glut1DS können sich an die Zentren für Seltene Erkrankungen wenden. Eine Liste mit spezialisierten Zentren und Ärztinnen und Ärzten hat der Glut1-Verein.
Glut1DS und Schwangerschaft?
Menschen mit Glut1DS können Nachwuchs zeugen. Die Wahrscheinlichkeit, dass das Kind eines Menschen mit Glut1DS und eines Menschen ohne Glut1DS ebenfalls Glut1DS bekommt ist 50:50. In der Fachliteratur gibt es eine Veröffentlichung von van der Louw et al. (2017) über eine Fallserie über Mütter, die sich auf Grund von Epilepsie ketogen ernähren und mögliche Einflüsse auf die Schwangerschaft. In einem weiteren Fachartikel von Kramer und Smith (2021) stellt einen Fallbericht einer Mutter mit Glut1DS dar, die schwanger wurde und deren Kind ebenfalls mit dem Glut1DS geboren wurde. Die Mutter ernährte sich auch während der Schwangerschaft ketogen und das Kind ab dem Tag der Geburt. Das Kind ist bisher altersgerecht entwickelt und ist symptomfrei.